

Newsletter November 2020
Die Digitalisierung der Medizin in Deutschland nimmt Fahrt auf und digitale Medizinprodukte, Software als Medizinprodukt oder Software für Medizinprodukte wird an Bedeutung gewinnen. Die Anforderungen dieser Art von Medizinprodukten haben wir in diesem Newsletter für Sie einmal zusammengetragen und listen Ihnen auch die ersten Digitalen Gesundheitsanwendungen auf.
Wie alle Medizinprodukte müssen hier die gesetzlichen Anforderungen erfüllt werden und hier gilt gerne der Vermutungscharakter: wenn eine harmonisierte Norm eingehalten wird, kann das als Nachweis gelten. Doch bedenken Sie das Ende der Übergangsfrist zur MDR im Mai 2021! Dann gelten besonders für Software strengere Anforderungen – insbesondere was die Klassifizierung betrifft (Regel 11). Doch auch für alle anderen Arten von Medizinprodukten wird sich etwas ändern. Aber verfallen Sie nicht in Panik und lassen Sie sich nicht entmutigen. Wir unterstützen und beraten Sie gerne bei allen Fragen rund um die Konformität zur MDR
Dr. Martin H. D. Neumann und Ihr MEC-ABC Team
Der Artikel
Im Oktober sind die ersten Digitalen Gesundheitsanwendungen in das Verzeichnis der erstattungsfähigen DiGA des BfArM aufgenommen worden. Diese Apps auf Rezept unterstützen bei der Erkennung und Behandlung von Krankheiten oder einer selbstbestimmten gesundheitsförderlichen Lebensführung und werden von den Krankenkassen erstattet. Das ist weltweit neu und einzigartig. Die ersten Apps, die in das Verzeichnis der erstattungsfähigen DiGA aufgenommen wurden, sind: Kalmeda (mynoise GmbH), eine App zur Unterstützung bei Tinnitus, Velibra (GAIA AG), eine App gegen Angststörungen, Somnio (mementor DE GmbH), eine App zur Behandlung von Ein- und Durchschlafstörungen, Zanadio (aidhere GmbH), eine App für die Behandlung von starkem Übergewicht und Vivira (Vivira Health Lab GmbH), eine App bei unspezifischen Rücken-, Knie- und Hüftschmerzen sowie Arthrose in Wirbelsäule, Knie und Hüfte.
Zurzeit befinden sich viele weitere DiGA im Prüfverfahren. Das DiGA Verzeichnis finden Sie auch hier: https://diga.bfarm.de/de/verzeichnis.
Die Digitalisierung der Medizin in Deutschland nimmt somit Fahrt auf. Digitale Gesundheitsanwendungen, Standalone Software, Health Software (als Medizinprodukt), Medizinproduktesoftware und Software as Medical Device (SaMD) sind Bezeichnungen für (digitale) Medizinprodukte deren Abgrenzung teilweise unscharf ist. Gemeinsam ist diesen Begriffen aber, dass das Herzstück des Medizinproduktes Software ist und diese Produktklasse in Zukunft an Bedeutung gewinnen wird. Passend zu dieser Entwicklung möchten wir Ihnen im November Newsletter eine Einführung zu den Anforderungen an Software als Medizinprodukt geben.
Anforderung an Software als Medizinprodukt
Das IMDRF (International Medical Device Regulators Forum) definiert “SaMD” als “Software, die für einen oder mehrere medizinische Zwecke verwendet werden soll, die diese Zwecke erfüllen, ohne Teil eines medizinischen Hardware-Geräts zu sein”. Eine wichtige Klarstellung hierbei ist, dass Software sich nicht auf den physischen Standort bezieht, von dem aus diese ausgeführt wird, sondern auf den regulatorischen Status der Software. Software kann auf universellen IT-Geräten, dem Smartphone/Tablet, in der „Cloud“ oder auch auf der Computerplattform eines medizinischen Hardware-Geräts ausgeführt werden. Wenn die Software aber zum Beispiel eine Hardware antreibt oder einen für das Hardwaregerät beanspruchten Zweck erfüllt, – also das medizinische Hardwaregerät benötigt die Software, um seinen beabsichtigten medizinischen Zweck zu erreichen – dann ist die Software kein SaMD, sondern Teil des medizinischen Geräts im regulatorischen Sinne.
Die EU verwendet anstelle von SaMD den Begriff „Medizinproduktesoftware“ (MDSW). Es definiert MDSW als Software, die allein oder in Kombination, für einen Zweck verwendet werden soll, welcher gemäß MDR der Definition von einem „Medizinprodukt“ entspricht. Die Qualifikation als MDSW ist unabhängig von ihrem Standort (z.B. Cloud, auf einem Computer, auf einem Mobiltelefon) und davon, ob die Software darüber hinaus auch die Verwendung von (Hardware-)Medizinprodukten steuert oder beeinflusst.
Die Geschichte der Entwicklung von Software als eigenständigem Bereich spiegelt sich auch in der Medizintechnik wider. Im Bereich der aktiven medizinischen Geräte wurde Software auch als Bestandteil eines medizinischen elektrischen Geräts angesehen. Die Anforderungen an ein solches programmierbares elektrisches Teilsystem (PESS) sind in der Norm IEC 60601-1 „Medizinische elektrische Geräte – Teil 1: Allgemeine Festlegungen für die Sicherheit einschließlich der wesentlichen Leistungsmerkmale“ beschrieben. Die zunehmende Komplexität von Software als Bestandteil eines Medizinprodukts oder zunehmend als eigenständiges Produkt führte zur
Erweiterung des Anwendungsbereichs und zur Veröffentlichung der Norm IEC 62304 „Medizingeräte-Software – Software-Lebenszyklus-Prozesse“.
Die IEC 62304 deckt den Software-Lebenszyklus von Medizinprodukten ab und ist eine zu empfehlende harmonisierte Norm, um gemäß der MDR EU 2017/745 dieser Forderung nach einem Software-Lebenszyklus zu begegnen. An dieser Stelle sollte erwähnt sein, dass zusätzlich die IEC 82304 „Gesundheitssoftware – Teil 1: Allgemeine Anforderungen für die Produktsicherheit“ beachtet werden sollte, da diese eine normative Lücke der IEC 62304 schließt. Die IEC 82304 gilt für Health Software, ob Medizinprodukt oder nicht und beachtet den gesamten Lebenszyklus inklusive der Produktvalidierung und -dokumentation, was bei der IEC 62304 nicht betrachtet wird. Allerdings empfiehlt sich für die Entwicklung und Verifizierung die Anwendung der IEC 62304.
Inhaltlich kann die IEC 62304 in vier grundlegende Prozesse unterteilt werden:
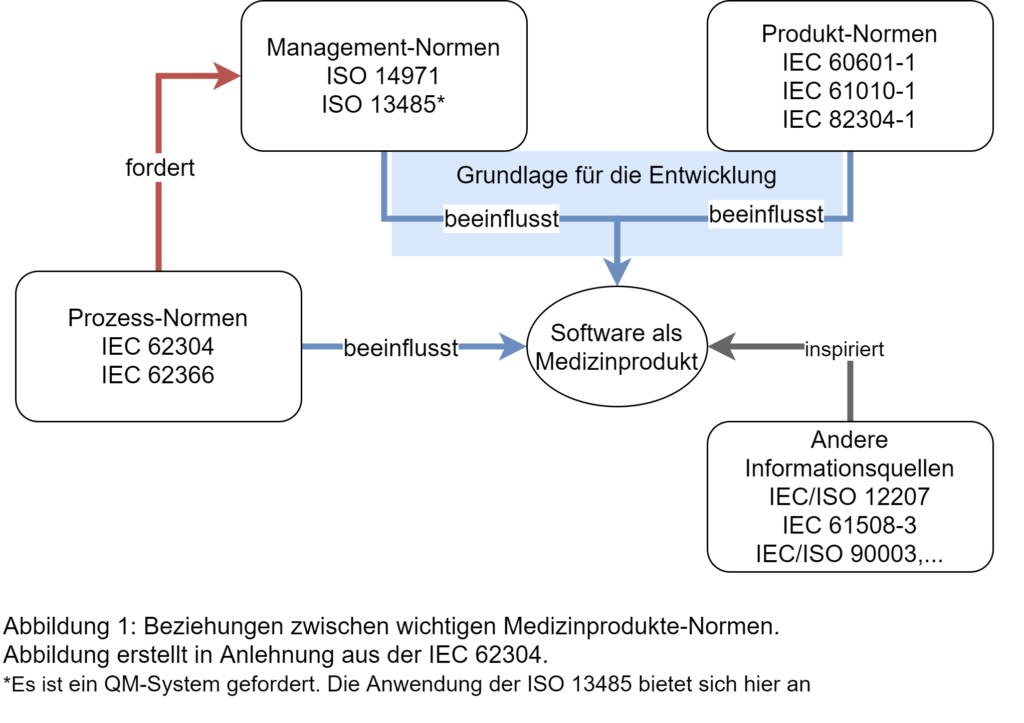
Die Anwendung der IEC 62304 setzt zudem voraus, dass bei der Entwicklung und Pflege der Software die Anforderungen eines QMS erfüllt werden. Es wird hierbei nicht direkt auf die ISO 13485 „Medizinprodukte – Qualitätsmanagementsysteme – Anforderungen für regulatorische Zwecke“ verwiesen, vielmehr soll der Hersteller die Fähigkeit ein Medizinprodukte nachhaltig und dauerhaft zu erstellen, beweisen. Dies kann durch ein QM-System unter Anwendung der ISO 13485 realisiert werden. Worauf die IEC 62304 allerdings explizit verweist und direkt darauf aufbaut, sind die Aktivitäten des Risikomanagements für Medizinprodukte gemäß der ISO 14971 „Medizinprodukte – Anwendung des Risikomanagements auf Medizinprodukte“. Abbildung 1 verweist auf die Beziehungen zwischen den oben genannten und anderen wichtigen Medizinprodukte-Normen im Kontext der IEC 62304.
Der Software Entwicklungsprozess startet mit der Planung. In der Planungs- und Entwicklungsphase sollten neben Angaben zu den Sicherheitsklassen auch folgende Informationen festgehalten werden
Entwicklungsprozess
Für den Entwicklungsprozess sollte zudem die wichtigste Aufgabe erfüllt sein: die Anforderungsanalyse. Die IEC 62304 setzt die Auseinandersetzung mit dem Gesamtsystem, Kundenbedürfnissen und weiteren Systemanforderungen voraus und baut auf diesen auf. Dementsprechend hat die Norm nicht die Zielsetzung, dass die Software dem Anwender den erwünschten Nutzen ermöglicht, sondern, dass das geforderte Software-System entsprechend der Erwartungen funktioniert. Hier können eine Vielzahl von Anforderungskategorien unterschieden werden, welche bei der Erstellung der Software-Spezifikation in Betracht gezogen werden sollten:
Risikomanagementprozess
n Risikomanagementprozess wird eine Gefährdungsanalyse durchgeführt. Im Mittelpunkt stehen Komponenten, die zu einer Gefährdungssituation beitragen könnten. Auch wenn Software als Ursache zu einer Gefährdungssituation beitragen kann, ist sie selbst keine Gefährdung. Zudem gilt zu bedenken, dass Softwarefehler immer systematische Fehler sind.
Als Ursache von Gefährdungssituationen kommen unter anderem die folgenden in Frage:
Diese Ursachen von Gefährdungssituationen gilt es zu analysieren. Die beteiligten Software-Komponenten als auch die Ursachen sollten in einer Risikomanagement-Akte dokumentiert werden. Anschließend gilt es passende Risikokontrollmaßnahmen zu definieren, zu implementieren und zu verifizieren/validieren. Dieser Prozess schließt also mit der Betrachtung von Maßnahmen zur Risikobeherrschung ab.
Es sollten zunächst gemäß der IEC 62304 die Sicherheitsklassen der Software nach einem 3-Klassenmodell charakterisiert werden (A= geringes Risiko, B=mittleres Risiko, C=hohes Risiko). Die Risikoklassen richten sich nach dem Beitrag der Software zu einer Gefährdungssituation. Die bereits erwähnten Prozesse sind abhängig von der jeweiligen Klasse. Hierbei stellt die IEC 62304 direkt Anforderungen an den Hersteller – abhängig von der Software-Sicherheitsklasse. So muss beispielsweise nur für die beiden höheren Sicherheitsklasse B und C das Design der Software Architektur[1] dokumentiert werden.
An dieser Stelle sollte angemerkt werden, dass grundsätzlich jedes Software-System einer Sicherheitsklasse zugeordnet werden muss und bis diese Zuordnung geschehen ist, gilt für das Software-System die Sicherheitsklasse C. Allerdings bietet die IEC 62304 die Möglichkeit, basierend auf der Aufteilung der Software-Komponenten, einzelnen Komponenten unterschiedliche Sicherheitsklassen zu geben. Dafür gilt, dass alle „Kind“-Komponenten die Software-Sicherheitsklasse der „Eltern“-Komponenten erben. Dokumentiert der Hersteller jedoch eine Begründung für die Klassifizierung in eine andere Software-Sicherheitsklasse, ist dies ebenfalls normenkonform.
Konfigurationsmanagement
Ein Konfigurationsmanagement wird für alle Sicherheitsklassen durchgeführt und hat sich zur Aufgabe gemacht, den (möglicherweise extrem hohen) Grad an Komplexität zu strukturieren und Veränderungen zu steuern und zu dokumentieren.
Das Konfigurationsmanagement dient unter anderem dazu, dass Hersteller sicherstellen, dass…
Die IEC 62304 fordert impliziert über den gesamten Software-Lebenszyklus zu gewährleisten, dass die Konfigurationselemente eindeutig identifizierbar und einem gegebenen Referenzpunkt zugeordnet werden können. Ein Konfigurationselement ist laut Norm eine „Einheit, die an einem gegebenen Referenzpunkt eindeutig identifiziert werden kann.“ Daraus kann man ableiten, dass alles, was während der Software-Entwicklung benötigt und erstellt wird, als Konfigurationselement angesehen werden kann. Darunter fallen unter anderen:
[1] Die Software-Architektur ist der Aufbau der Software unter der sichtbaren Oberfläche. Man unterscheidet Software-Architektur (Marko, den Aufbau im Großen) und Software-Design (Mikro, den Aufbau im Kleinen).
Wartung und Problemlösung
Sobald die Freigabe der Software erfolgt ist, gilt die Entwicklung als abgeschlossen. Hierauf folgen übergeordnete Prozesse, Aufgaben und Aktivitäten wie beispielsweise die Validierung. Die Freigabe und die Validierung werden nicht von der IEC 62304 abgedeckt – hierfür kann die regulatorische Lücke mit Hilfe der IEC 82304 geschlossen werden. Trotzdem sind die Aktivitäten der IEC 62304 mit der Entwicklung der Software nicht beendet, denn es können über den Lebenszyklus der Software Probleme auftauchen, welche aufgrund von inkompatiblen Schnittstellen, neu identifizierten Schwachstellen in Sicherheit oder Funktionen entstehen, oder die unter bestimmten Voraussetzungen nicht das erwartete Ergebnis liefern. Dementsprechend müssen Rückmeldungen zur entwickelten Software überwacht werden.
Vom Hersteller wird gefordert, die Aktivitäten bezogen auf die Software-Wartung festzulegen und zu planen. Die Planung muss Folgendes adressieren:
Schließlich fordert die IEC 62304 Vigilanz und einen Problemlösungsprozess. Hierbei sollen Probleme, die im Zusammenhang mit der Anwendung medizinischer Software auftreten, berichtet, untersucht und beteiligte Stellen unterrichtet werden. Dabei ist es egal, ob das Problem innerhalb oder außerhalb der Organisation, während der Software-Entwicklung oder nach der Software-Freigabe identifiziert wurde. Alle Probleme sollten gleichermaßen behandelt werden. Dabei sollte das Problem untersucht und mögliche Ursachen und Abhängigkeiten identifiziert werden. Anschließend folgt die Analyse: Betrifft das Problem auch andere Systeme oder Schnittstellen? Unter Verwendung des Software-Risikomanagement-Prozesses sollte die Relevanz und Auswirkung des Problems auf Leistungsfähigkeit, Sicherheit oder Datensicherheit der Software evaluiert werden.
Wichtiger Hinweis
Bitte beachten Sie unseren wichtigen Hinweis: Aufgrund der COVID19-Pandemie wurde die Übergangsfrist der MDR auf den 26. Mai 2021 verschoben. Bei vielen Herstellern hat das leider nicht dazu geführt, dass sie die Zeit für die Umstellung auf die MDR nutzen, sondern eine (Re-)Zertifizierung nach den alten Richtlinien MDD/AIMD angestrebt haben und dies auch ein halbes Jahr vor Ende der Übergangsfrist noch tun. Doch auch für bereits im Markt befindliche MDD/AIMD-zertifizierte sowie neu unter MDD/AIMD zuzulassende Produkte schreibt Artikel 120 der MDR („Übergangsbestimmungen“) vor, dass Hersteller bis zum Ende der Übergangsfrist die „vorliegenden Anforderungen an die Überwachung nach dem Inverkehrbringen [Post-Market Surveillance, PMS], die Marktüberwachung, die Vigilanz, die Registrierung von Wirtschaftsakteuren und von Produkten“ nach MDR einführen müssen. Dazu kommt, dass MDD/AIMD-Zertifikate spätestens am 27. Mai 2024 ihre Gültigkeit verlieren und die letzten Produkte auf dem Markt bis 27. Mai 2025 abverkauft werden müssen. Sie können die Umsetzung der kompletten MDR also nur noch für kurze Zeit aufschieben, aber nicht aufhalten! Stellen Sie lieber jetzt auf die MDR um, wir begleiten Sie gerne.

virtual.MEDICA startet in Kürze
Bald findet die virtual.MEDICA statt (16.11.-19.11.2020, die Chatfunktion im Rahmen der virtual.MEDICA steht bis zum Mai 2021 zur Verfügung).
Wir werden zwei Websessions anbieten:
16.11.2020 – 14:00-14:30 Uhr –
Klinische Daten in der MDR: Klinische Bewertung, PMCF & Co.
17.11.2020 – 12:00-12:30 Uhr –
Digitale Gesundheitsanwendungen (DiGA): Die App auf Rezept
Sie möchten sich mit uns in Videomeetings austauschen? Dann nehmen Sie am Matchmaking der MEDICA teil! Kommen Sie (virtuell) vorbei, wir freuen uns auf Sie!

Digitale Gesundheitsanwendungen
Die MEC//ABC GmbH ist jetzt Fördermitglied des Spitzenverbandes Digitale Gesundheitsversorgung (www.digitalversorgt.de)! Wir freuen uns sehr über die Aufnahme, denn auch wir wollen proaktiv die Digitalisierung in der Medizin unterstützen, uns in der Verbandsarbeit einbringen und mit unserem Know-How den SVDGV und seine Mitglieder mit Rat und Tat in Sachen MDR-Konformität, klinischer Bewertung, Risikomanagement, und Usability Engineering unterstützen!
Aufnahme in das Verzeichnis für Digitale Gesundheitsanwendungen: Online-Einführungsseminar
Wir unterstützen Sie auch im Bereich Digitale Gesundheitsanwendungen (DiGA)-Verfahren. Ordern Sie doch noch heute unser kostenfreies Online-Seminar zum Einstieg in das Thema DiGA und das Antragsverfahren über: info@mec-abc.de.
Chat-Funktion
Zusammen mit dem NovemberNewsletter startet auch ab dem 01.11.20 unsere Chatfunktion auf unserer Website( www.mec-abc.de).
Hier können Sie persönlich mit einem Experten der MEC//ABC in den Kernzeiten von 9-11 Uhr chatten (Analyse der Traffic-Zeiten permanent, ggf. dann Anpassung der Chatzeiten nach der Startphase).
Youtube
Besuchen Sie unseren neuen Youtube-Channel:
MEC ABC Youtube Kanal
Dort finden Sie hilfreiche Informationen in Form von anschaulichen Cartoons, Q&A Sessions und Erklärvideos.
Sie sehen gerade einen Platzhalterinhalt von Vimeo. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von YouTube. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie müssen den Inhalt von reCAPTCHA laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Facebook. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Instagram. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Google Maps. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie müssen den Inhalt von hCaptcha laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie müssen den Inhalt von reCAPTCHA laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Turnstile. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von X. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr Informationen