

Newsletter Mai 2021
Guten Tag liebe Leserinnen und Leser,
vor Kurzem haben Frau Deserno und ich ein full scope 13485 QM-Audit remote durchgeführt. Dies war ungleich anstrengender als ein Vorort-Audit. Was aber wirklich schön war, war die Verzahnung vom Clinical Part mit dem QM-System. Zu sehen, zu hören und zu erleben, wie die Schnittstellen auch gelebt werden.
Die klinische Bewertung, die klinischen Prüfungen und auch das Risikomanagement waren Teil des QMS – was noch nicht bei allen unserer Kunden der Fall ist. Dies liegt oft daran, dass die Ressourcen nicht verbraucht werden sollen oder die Qualitätskriterien für diese Bereiche nicht klar sind.
In unserem Newsletter finden Sie einige Anregungen, wie Sie die Anforderungen zum Clinical Part für Legacy Device erfüllen können.
Viel Spaß beim Lesen und bleiben Sie gesund!
Dr. Nadine Leistner und das Team der MEC-ABC GmbH

Wie in der MDR beschrieben, müssen für Legacy Devices ausreichend klinische Daten vorliegen, damit Sie bei der Zertifizierung nicht klinisch geprüft werden müssen.
Wir haben bei einigen Kunden erlebt, dass das auch für Produkte, die schon mehrere Jahrzehnte auf dem Markt sind und als sicher einzustufen sind, gilt und von den benannten Stellen eingefordert wird.
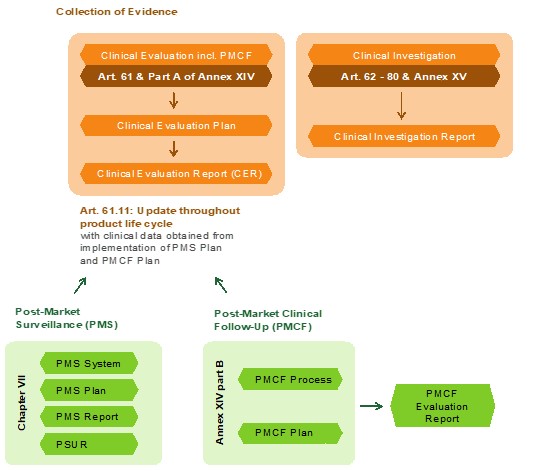
Schnittstellen bei klinischen Daten
Zentrales Dokument für den Beleg der vorhandenen klinischen Daten ist die klinische Bewertung nach Anhang XIV der MDR.
Hierzu sind in der MDR viele Schnittstellen deutlicher gemacht worden. Beim Risikomanagement wird beschrieben, dass dieses mit den Verfahren der klinischen Bewertung verknüpft sein soll und regelmäßig aktualisiert werden soll. Bei der Marktüberwachung ist neben der reaktiven Form der Post Market Surveillance auch die proaktive Sammlung von klinischen Daten das Post Market Clinical Follow-up hinzugekommen. Es ist ein komplexes Berichtswesen gefordert, welche abhängig von der Risikoklasse ist.
Die Möglichkeiten die Herausforderungen der Umstellung der MDR im Unternehmen zu meistern sind vielfältig. Für den Top-Down-Ansatz sollten Sie als erstes über die benötigten Ressourcen entscheiden und diese freigeben. Denn der Aufwand ist auf jeden Fall größer als vorher. Wenn Ihnen Expertise im Haus fehlt, versuchen Sie Experten einzustellen oder suchen sich einen verlässlichen Dienstleistungspartner. Viele Aufgaben lassen sich digitalisieren, wie die Aktenführung der technischen Dokumentation oder die Datensammlung für das PMCF.
Überlegen Sie sich aus betriebswirtschaftlicher Sicht, welche Produkte sich mit den Aufwänden finanziell lohnen und welche nicht. Sortieren Sie dabei auch die vorhandenen Produkte in sinnvolle Produktfamilien.
Wenn Sie sicher sind, dass Sie klinisch prüfen müssen, kümmern Sie sich frühzeitig um Ihre Qualifikation als Sponsor einer klinischen Prüfung. Zudem bleiben Sie in Kontakt mit Prüfärzten bzw. -zentren. Sie sollten dafür sorgen, dass die Prüfarzte die geforderten Qualifikationen zur klinischen Prüfung von Medizinprodukten haben oder dafür sorgen, dass diese in Zukunft erfüllt sind.
Aufgrund der beschriebenen Schnittstellen sollten Sie auch Ihr Qualitätsmanagement anpassen.
Für jedes Produkt, welches Sie in Zukunft weiterführen wollen, sollten Sie eine eingehende Analyse durchführen. Bestimmen Sie als aller erstes die wichtigsten „regulatorischen Eckdaten“. Dies sind Zweckbestimmung, Indikation, Target Population und Medical Claims.
Diese Daten benötigen Sie, um Ihre Produkte auf MDR umzustellen. Die Indikation und die Target Population scheinen bei einigen Produkten nicht so leicht bestimmbar zu sein. Jedoch auch hier ist dies unerlässlich, um den Anforderungen der MDR zu genügen.
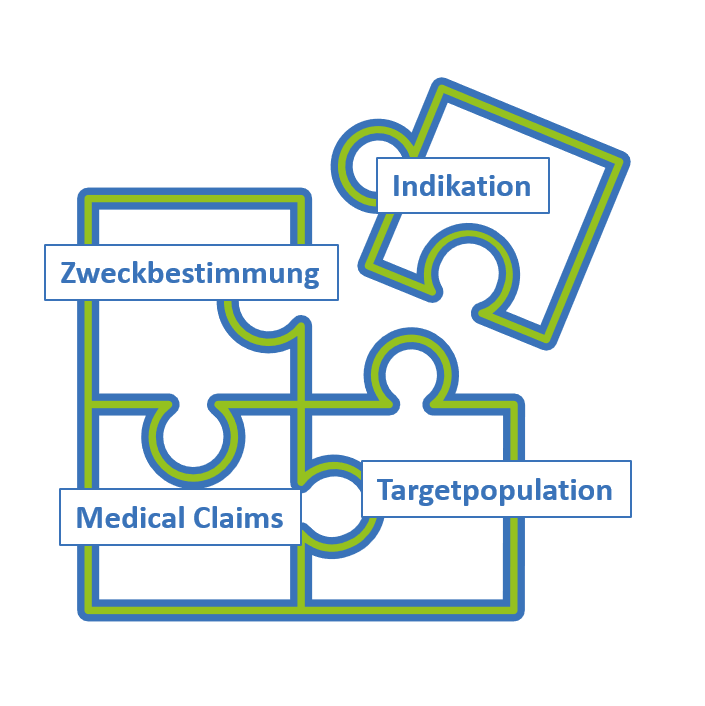
Die regulatorischen Eckdaten sollten mit in die klinische Bewertung einfließen (genauso wie die Ergebnisse der Risikoanalyse). In der klinischen Bewertung wird dann jedes einzelne Puzzle-Stück betrachtet. Wobei auch auf dem Weg rauskommen kann, dass sich einige Indikationen oder Targetpopulationen betriebswirtschaftlich nicht lohnen. Oder es kann klar werden, dass einige Medical Claims nicht haltbar sind.
Wenn die klinische Bewertung abgeschlossen ist, wird diese (wenn gut ausgeführt) fehlende Daten aufzeigen. Beispielsweise, wenn Daten in einer Indikation oder einer Population noch nach zu erheben sind.
Die Sammlung der klinischen Daten wird von verschiedenen Einflussgrößen bestimmt. Es sollten die Vorgaben aus der klinischen Bewertung umgesetzt werden (Claims, State-of-the-Art, Risikoklasse, Evidenzklasse). Eine weitere Komponente dabei ist jedoch die rechtliche Einordnung Ihrer PMCF-Aktivität, die in einigen Fällen eine klinische Studie ist. Bei dieser sollten Sie alle Eingabegrößen betrachten und sich wenn nötig beraten lassen. Faktoren sind beispielsweise, ob mit pseudonymen oder anonymen Daten gearbeitet wird, oder eine zusätzliche Strahlenbelastung auftritt.
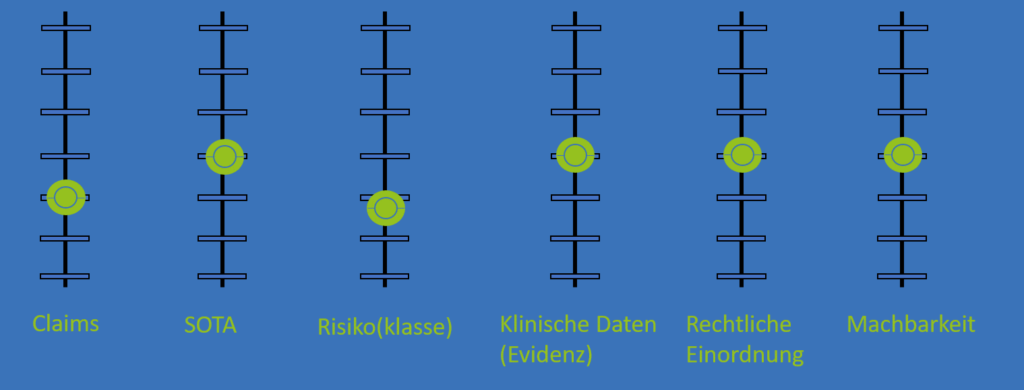
Und eine wichtige, wenn nicht die wichtigste beim Erfolg der Aktivität ist die Machbarkeit der Datenerhebung. Es stehen Fragen im Vordergrund wie: Wo werden die Daten erhoben? Können diese überhaupt erhoben werden? Passt die Datensammlung/klinische Studie überhaupt zu den Abläufen in der Klinik oder der Patienten?
Wenn alle Stellschrauben bedient worden sind, kommt am Ende ein Plan heraus, der Ihnen und der benannten Stelle aufzeigt, wie eventuelle Gaps für die klinischen Daten Ihrer Produkte geschlossen werden können.
Wichtig ist es, bei der Umsetzung der MDR nicht das große Ganze aus den Augen zu verlieren. Denn alle Einzelteile der technischen Dokumentation müssen stimmig zusammenpassen und können nicht mehr isoliert betrachtet werden. Hierzu gehören ebenfalls regelmäßige, geplante Updates.
Stellen Sie sich den Herausforderungen, denn Sie sind der Spezialist für Ihr Produkt!
Wenn Sie Expertise oder Unterstützung benötigen, kontaktieren Sie mich gerne unter 0241/ 519 674 86-01 oder schreiben Sie mir N.Leistner@mec-abc.de
Sie sehen gerade einen Platzhalterinhalt von Vimeo. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von YouTube. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie müssen den Inhalt von reCAPTCHA laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Facebook. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Instagram. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Google Maps. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie müssen den Inhalt von hCaptcha laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie müssen den Inhalt von reCAPTCHA laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Turnstile. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von X. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr Informationen