

Newsletter September 2020
Unsere Erfahrungen bei der Erstellung Klinischer Bewertungen haben gezeigt, dass eine Analyse der Technischen Dokumentation maßgeblich für den Erfolg der Zertifizierung, auch des Clinical Parts, ist.
Mit Hilfe unserer Gap Analyse zeigen wir Ihnen eventuelle Unstimmigkeiten und erforderliche Entscheidungen in der Technischen Dokumentation auf. Dadurch bekommen Sie als Hersteller einen ersten Überblick über den Stand Ihres Medizinproduktes im Hinblick auf die Anforderungen an klinische Daten der MDR.
Worauf wir achten, wenn wir Ihre bestehende klinische Bewertungen analysieren und eine Konzept für die klinischen Daten erarbeiten, können Sie im Artikel lesen.
Am 2. Oktober um 10 Uhr findet unser kostenloses Online-Seminar zum Thema Fast-Track-Verfahren der Digitalen Gesundheitsanwendung statt.
Zudem gibt es erste Ideen für eine Antragsstellung für den Covid-19 Call des BMBF’s im Bereich Unterbrechung der Infektionsketten in sensiblen Gesundheitseinrichtungen. Melden Sie sich gerne, wenn Sie dazu mehr erfahren möchten.
Dr. Max Woitok und Ihr MEC-ABC Team
Die Artikel
In den zahlreichen Beratungsgesprächen mit unseren Kunden tauchen immer wieder Fragen auf, welche Informationen und Daten für eine Klinische Bewertung benötigt werden.
Die klinische Bewertung ist systematischer und geplanter Prozess zur kontinuierlichen Erzeugung, Erhebung, Analyse und Bewertung klinischer Daten zu einem Produkt ist, um die Sicherheit und Leistungsfähigkeit, einschließlich des klinischen Nutzens, des Produkts bei der vom Hersteller vorgesehenen Verwendung zu überprüfen.
Dieser Prozess ist dabei ein Zusammenwirken zwischen den Dokumenten der Technischen Dokumentation, des Riskmanagements und Qualitätsmanagements, der Gebrauchstauglichkeit sowie des Klinischen Bewertungsplans (Clinical Evaluation Plan, CEP) und Bewertungsreports (Clinical Evaluation Report, CER), sowie der Überwachung nach der Inverkehrbringung (Post-Market Surveillance (PMS) und Post-Market Clinical Follow-up (PMCF)) des Medizinproduktes.
Neue Erkenntnisse über das Medizinprodukt haben damit direkten Einfluss auf alle der genannten Punkte in dem Prozess der Klinischen Bewertung.
Unabhängig vom Medizinprodukt sind bestimme Informationen wesentlich für die Erstellung eines CEP und des CER, diese Informationen nennen wir regulatorische Eckdaten. Auf diese regulatorischen Eckdaten möchten wir in diesem Newsletter im Detail eingehen. Diese Eckdaten sollten vor einer Klinischen Bewertung durch den Hersteller festgelegt werden, können aber auch noch während der Erstellung der Klinischen Bewertung verändert werden.
Die genaue Formulierung z.B. der Zweckbestimmung entscheidet dabei unter anderem über Risikoklasse des Medizinproduktes, den Umfang der Medizinischen Angaben (Medical Claims) und hat damit direkten Einfluss auf das PMCF, das ein verpflichtender Bestandteil der PMS ist. Aber was genau sind diese regulatorischen Eckdaten?
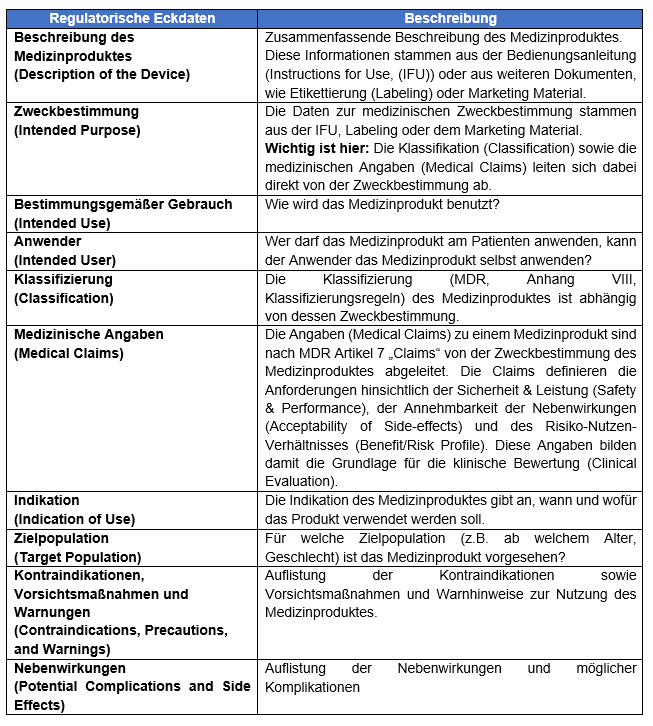
Warum sind diese Daten so wichtig für Sie intern aber auch zur Erstellung der Klinischen Bewertung für uns? Die folgende Tabelle dient hier als Überblick:
Table: Regulatorische Eckdaten zur Erstellung einer Klinischen Bewertung
Zur Erfüllung aller der Anforderungen der MDR für eine Klinische Bewertung werden in regelmäßigen Abständen Leitfäden der Medical Device Coordination Group (MDCG) der EU-Kommission herausgegeben. Diese haben wir Ihnen in unserem ersten Newsletter im August vorgestellt.
Wir wissen, dass die Klinische Bewertungen mit allen benötigten Anforderungen eine große Herausforderung ist. Die Erstellung der Dokumente bedingt zeitliche und personelle Kapazitäten in Ihrem Team.
Hier unterstützen wir Sie mit unseren Fachkenntnissen und helfen bei der Erstellung und regelmäßige Überarbeitung der Klinischen Bewertung. Schreiben Sie uns an! Info@mec-abc.de
Aktuelles aus der MEC-ABC
(Remote)-Team als Service Die Herausforderungen an den Clinical Part der Zertifizierung der MDR sind komplex. Deswegen bietet die MEC-ABC Ihnen auch an Sie als (Remote)-Team als Service zu unterstützen. So bekommen Sie für einen definierten Zeitraum die benötigten Ressourcen und Kompetenzen, um die entscheidenden Weichen für Ihre Produkte zu stellen. Wir bereiten uns auf die MEDICA (11.-19.11.2020) vor Unter Beachtung aller Vorgaben des Hygiene- und Infektionsschutzkonzeptes der MEDICA finden Sie unseren Stand am Landsgemeinschaftsstand des Landes Nordrhein-Westfalen. Kommen Sie vorbei, wir freuen uns auf Sie! DiGA-Verfahren In Zukunft werden wir Sie auch im Bereich DiGA-Verfahren unterstützen und laden Sie am 2. Oktober 2020 zu unserem 1 Std. Online-Seminar ein. Wir werden Ihnen das Verfahren und die mögliche Unterstützung vorstellen. Zur Anmeldung senden Sie einfach eine Email an Info@mec-abc.de
Hier finden Sie auch einen brandaktuellen Statusbericht zum DiGA Verfahren
DiGA Antragsverfahren – die ersten 100 Tage
Bekanntmachungen und Hinweise
Der Beuth Verlag stellt Normen für Covid-19 bereit:
https://www.beuth.de/de/sonderausgaben-normen-medizinische-ausruestung
Richtlinie zur Förderung von Projekten zum Thema „Prävention und Versorgung epidemisch auftretender Infektionen mit innovativer Medizintechnik“
https://www.bmbf.de/foerderungen/bekanntmachung-3100.html
Richtlinie zur Förderung von internationalen Verbundvorhaben in Wissenschaft und Forschung zwischen Südostasien und Europa mit dem Themenschwerpunkt Infektionsforschung im Rahmen des Southeast Asia-Europe Joint Funding Scheme
https://www.bmbf.de/foerderungen/bekanntmachung-3112.html
BAFA, „Bundesförderung von Produktionsanlagen für Schutzausrüstung“: https://www.foerderdatenbank.de/FDB/Content/DE/Foerderprogramm/Bund/BMWi/bundesfoerderung-von-produktionsanlagen-von-perso.html
BfArM, Hinweise für Hersteller, Importeure und Vertreiber zur Sonderzulassung von medizinischen Gesichtsmasken sowie partikelfiltrierenden Halbmasken (FFP2 und FFP3)
Sie sehen gerade einen Platzhalterinhalt von Vimeo. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von YouTube. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie müssen den Inhalt von reCAPTCHA laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Facebook. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Instagram. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Google Maps. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie müssen den Inhalt von hCaptcha laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie müssen den Inhalt von reCAPTCHA laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Turnstile. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von X. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr Informationen